Results of VEGA:
A Tutorial For An Adequate Interpretation
Introduction
VEGA and ToxRead are tools supporting the evaluation of the properties of chemical substances. The user is the ultimate responsible for this evaluation. The user should be aware of what these tools can provide, and what they cannot.
VEGA offers a broad series of information, and the user should be aware of the elements offered by VEGA to assess a chemical substance. Additional inputs may be also obtained by a joint use of VEGA and ToxRead. Considering all the evidences provided by VEGA the user may evaluate how credible is a prediction, and the level of uncertainty associated to the prediction.
VEGA is a complex platform which basically includes a number of QSAR models, and an independent tool helping the user in the evaluation of the result, through the Applicability Domain Index. In this way VEGA combines QSAR and read across tools. The QSAR prediction models derive from CAESAR, T.E.S.T., SARpy, EPISuite, Toxtree, and other tools. New models are frequently added. Registered users are notified when new models are available.
A completely independent algorithm is used to assess the reliability of the model prediction through what we call the Applicability Domain Index. This algorithm works on all the separate QSAR models. This algorithm shows similar compounds, assesses the QSAR results on the similar compounds, and analyzes some relevant chemical features in the target compound and its related compounds.
An automatic evaluation for read-across is done, and thus this tool can be used for read across, independently from the prediction obtained through the QSAR models. In the ideal situation both pieces of information, from the QSAR model and from the Applicability Domain Index, should be assessed. The user is the ultimate responsible of the assessment, but we work in the direction to offer most of the information to the users, for a reproducible assessment.
For the evaluation of the results, we recommend to take advantage of both these tools, QSAR and read across. The user should analyze the results shown on the similar compounds, and the additional documentation provided on the occurrence of relevant chemical moieties.
In the frequent case of more than one model available for the same endpoint, the assessment should be done independently on each model, and then an overall assessment should integrate the results from each model.
Below we describe the unique Applicability Domain Index implemented within VEGA, we show how to read and interpret the information from the VEGA report, and how to take advantage on the information relative to the structural alerts.
The Applicability Domain Index
For a certain endpoint and model, the user will get the prediction form the QSAR model, and a number of elements to evaluate the prediction. These elements are provided by VEGA with a program working independently from the QSAR model.
Any QSAR model is based on three pillars: the property to be studied (for instance mutagenicity), the chemical information, and the function linking the property and the chemical. Thus, to evaluate the prediction, we developed parameters which refer to each of these three components of the conceptual QSAR models: the property, the chemical information, and the algorithm. These parameters are then merged into a single value, called applicability domain index (ADI).
Other programs address the applicability domain in a qualitative way (in or out), some also in a quantitative way with a numerical value, most refers to the chemical similarity only, but the tool developed within VEGA is unique since it conceptually refers to all the pillars of the QSAR model: the chemical information, the property, and the algorithm linking the chemical and the property.
The ADI is a guidance for the user, but it cannot be used in an uncritical way. Of course, within a strategy of prioritization, and screening of large series of substances, the ADI can be applied as a first filter, and the manual assessment described below cannot be applied for large numbers of substances. Indeed, QSAR models can be used for screening, or for the assessment of a single substance, and the role of the user is much more important in the second case.
We have noticed that the use of ADI is useful to improve the reliability of the predictions, working on collections of substances, for different endpoints. See for instance the final report of the CALEIDOS project.
However, the user may disagree on the prediction, or on the relevance of certain score indicated within the ADI. Below we provide some examples, to better explain the use of all the pieces of information provided by VEGA, and what the user should do.
The elements to be evaluated
The prediction of the QSAR model. This is produced by the QSAR model. The use of more models, if available, is recommended. If they agree, this is a reason for higher overall reliability. If they disagree, the reason should be addressed. The elements offered by the ADI and the examples below should drive this process.
The similarity of the related compounds. This relates to the chemical information of the model. Similarity depends on many factors, and there is no absolute measurement for it. We optimized the algorithm of similarity used in VEGA on the basis of a check with 4 millions compounds, and this is an advantage of VEGA compared with other programs. However, the final evaluation on similarity should be done by the user, as shown below, and the user may discard some substances. See for instance the case discussed below on the use of the information based on the structural alert.
The software calculates how similar is the similar compound providing a score, between 1 (in case of identity) and 0.
Usually values lower than 0.75 indicate that the similar compound has important differences compared to the target.
There is no threshold, but a guidance value, reported in the information document available for each model within the VEGA platform. This value depends on the number of chemicals in the training set, and thus it may vary. The similarity is calculated as described (http://jcheminf.springeropen.com/articles/10.1186/s13321-014-0039-1). The user should always check manually the similarity between the target and the similar compound. Indeed, there is no perfect similarity algorithm, and the similarity concept itself is arbitrary. The assessment within VEGA has a chemical meaning, but further check should be done based on particular chemical features which are related to the specific endpoint, and are addressed in VEGA in an automatic way through the independent parameters described below. Thus, the user should take into account other chemical features specific for the endpoint, in particular through the analysis of the structural alerts. Some QSAR models are based on about ten thousands compounds, and in this case it is easy to find similar compounds. However, for other properties the available experimental values are much less, and thus the use of the similarity information is reduced.
The similar compounds can be used also within a read across perspective. Indeed, VEGA combines QSAR and read across, but the user may decide to use only the QSAR result or the read across approach. As much as possible we recommend using both approaches, within a weight-of-evidence strategy. This reinforces the assessment. If the similar compounds are not so similar, it is inappropriate to use read across.
The presence of unusual fragments. This relates to the chemical information of the model. VEGA identifies the presence of rare fragments, not common in the set of compounds at the basis of the specific model. Thus, this factor identifies a lack of knowledge on a component present in the molecule.
The check of the descriptor range. This relates to the information on the algorithm of the model. VEGA evaluates if the descriptors of the target compound have values in the range of those related to the substances in the training set. Also the molecular weight is considered, if it is inside the range of values of those of the training set.
The sensitivity analysis of the descriptors. This relates to the information on the algorithm of the model. VEGA evaluates if a change of 10% of the descriptor values of the target compound provoke a large variation of the predicted value. This indicates an area of higher uncertainty of the model, associated to possible activity cliffs. This algorithm is quite unique of VEGA.
The concordance between the predicted value of the target compound, and the experimental values of the similar compounds. This relates to the information on the toxicity/property value. The experimental values can be used alone, without the predicted values, and thus, in practice, it can be used for read across. If there is agreement between the predicted value and the experimental values of the similar compounds, this is the ideal situation. If there is disagreement, the user should decide if there are sufficient elements to take a decision. The user may disregard part of the information, if the remaining information is sufficient and convincing. In this process, the user should carefully evaluate the reasons for the effect, also considering the eventual presence of structural alerts, as below explained.
This parameter, the concordance, is the most critical one. All the other parameters can only affect the reliability of the assessment, and indicate uncertainty in the prediction. However, considering the experimental values of the similar compounds the user has novel information, from a different source, not from the QSAR model.
The accuracy of the prediction. This relates to the information on the toxicity/property value. This parameter indicate if the specific VEGA model face problems in the prediction of similar compounds, and thus if in the local area of the target compound there may be issues.
The maximum error in prediction. This relates to the information on the toxicity/property value. In case of models for continuous values, VEGA provides this parameter, which may indicate high level of uncertainty.
The presence of structural alerts. This relates to the information on the toxicity/property value. There are structural alerts (SA) indicated by VEGA, and also by ToxRead. These SA are of different nature.
- SA associated to a positive effect, such as genotoxicity.
- SA which are exception rules of the previous SA.
- SA which reduce the positive effect, in a generic way. While the previous rules block the effect provoked by the SA, these SA modulate the effect, and may also cancel the effect, because a parallel process may prevail.
- SA which are neutral (these may be called rules, instead than SA, due to the lack of effect). The interest on the information about these SA is that the user may want to analyse the possible reason of effect in a certain molecule; if a certain fragment is known to be non-toxic, it may be skipped form the evaluation.
The SA can be associated to known plausible effect. In several cases there are papers explaining the possible mechanism, which may also apply to the chemical under investigation. There are level of uncertainty associated to these SA as well. The user should not consider the presence of an alert of this kind as a demonstration of the effect. Indeed, most of these SA do not have an accuracy of 100%, and those with this level of accuracy have a limited number of chemicals. In other terms, there are substances with SA of this kind which are not positive.
These SA are represented with different structures in the different collections of SA. In other terms, there is not an agreement on which are the active SA.
There are different levels of accuracy depending on the “sub-families” of SA. In other terms, is the percentage of positive chemicals associated to aromatic amines for genotoxicity, for instance, is X %, it may be higher for a subset of aromatic amines. Conversely, it may be reduced for other subsets. It is convenient to look for the most specific alert, which better fit the structure of the substance of interest. ToxRead contains the largest list of these SA for mutagenicity.
There are different methods to derive SA. In VEGA and ToxRead there are collections of SA derived by studies done by human experts, like those present in Toxtree, the so-called Benigni-Bossa rules for genotoxicity. These SA are also present in VEGA and ToxRead. There are SA derived by human experts of Mario Negri (E. Benfenati et al., 2015 ), present in ToxRead. There are SA derived from computer programs, like SARpy, developed by Politecnico di Milano, or by another program developed by CRS4. All these SA contain both active and inactive fragments, with the exception of those from Toxtree.
The occurrence of concordant structural alerts in the similar compounds. This relates to the information on the toxicity/property value. This information is associated to the information on the concordance above discussed. This information is important to decide if the similar compound is relevant or not. If the similar compound has the same SA of the target compound, the similar compound is relevant. Of course, this applies only in the case that the effect can be explained by the SA, thus it does not apply to all cases. Even in cases in which there is the same SA in both the similar and the target compound, the user should evaluate if there are reasons to modulate or cancel the positive effect. See above, the SA which are exception rules, or modulate the effect. For more details see below.
The VEGA report (PDF version)
VEGA provides the user with all the information regarding the prediction and on how to interpret it.
The report is organized in the following sections*:

* Currently the report generated by VEGA contains the described sections but they are not yet organized with this layout, we will update this soon.
General assessment of the results of the VEGA models: The BCF case Study
In the case of BCF the documentation in the VEGA report also involves the analysis of the logP values. The description on the BCF and logP values for the target compound and for known chemicals, as given in the Possible use and uncertainty section of the VEGA report, should be evaluated as well.
For simplicity’s sake we can identify two situations:
Situations:
Reliable Evaluation
The general VEGA evaluation of the applicability domain is good: the ADI is > 0.85 and a green button is shown in the summary evaluation
This means that all automatic checks gave good results, as detailed within the Applicability Domain section of the Report.
The user should anyhow check the occurrence of unusual findings. In particular, within the Applicability domain: Similar compounds, with predicted and experimental values section the similar compounds should be visually analyzed: chemicals should be similar and the experimental values of the most similar compounds in agreement with the predicted one. Furthermore, the predicted BCF value should not be an outlier within the figures of the Reasoning: Analysis of molecular descriptors section.
Examples
NAME
1,3-diisopropylbenzene
CAS
99-62-7
SMILES
c1cc(cc(c1)C(C)C)C(C)C
PREDICTED VALUE
3.03
Model assessment
 Prediction is logBCF = 3.03, the compound is IN the applicability domain and prediction seems to be reliable.
Prediction is logBCF = 3.03, the compound is IN the applicability domain and prediction seems to be reliable.
Anyway some issues could be not optimal:
- similar molecules found in the training set have experimental values that slightly disagree with the target compound predicted value.
The following relevant fragments have been found: Thiobenzene residue (SR 04); SO3H group (PG 02).
Let’s consider the read across evaluation:
- There are chemicals with very good similarity. Look at the similarity values > 0.95 for three of them, and at the chemical structures.
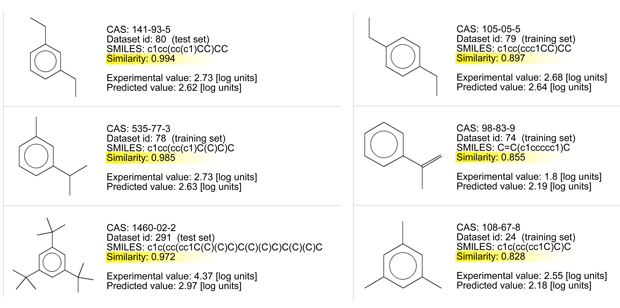
- All of them have an aromatic ring and short aliphatic substituents.
- Figure 1 shows that the target chemical is within the typical relationship between logP and BCF.
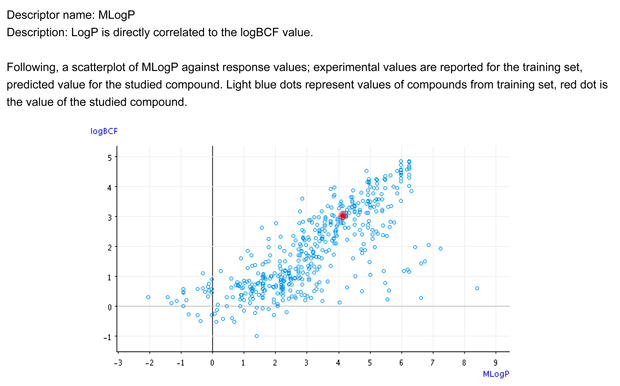
- Figure 2 shows that the BCF and logP values of the target compound are close to the values of the similar compounds. The similar compound is inside the BCF and logP values of the similar compounds.
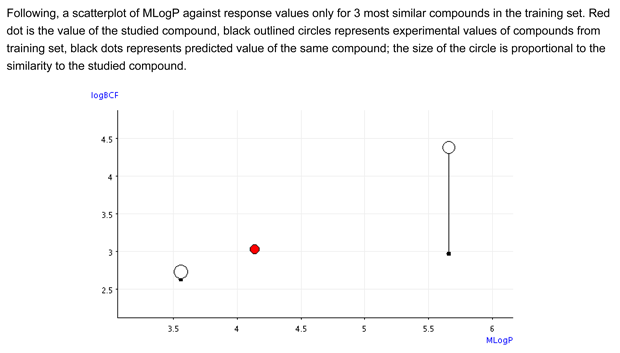
Conclusion: The predicted value is reliable and the tool in support to read across confirms this.
NAME
p-Toluidine-o-sulfonic acid
CAS
88-44-8
SMILES
O=S(=O)(O)c1cc(ccc1(N))C
PREDICTED VALUE
0.21
Model assessment
Prediction is logBCF = 0.21, the compound is
IN the applicability domain and prediction seems to be reliable.
Let’s consider the read across evaluation:
- There are chemicals with good similarity. Look at the similarity values > 0.75, and at the chemical structures.
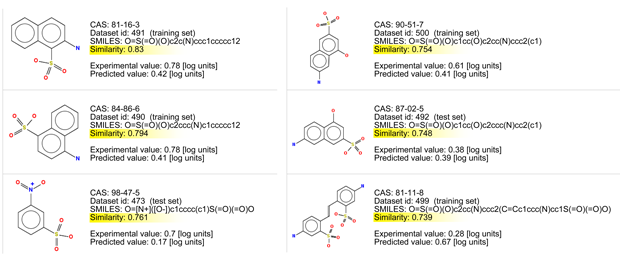
- In particular two of them are quite similar, and have an aromatic nucleus, and the same two polar residues: sulfonic acid, and primary amine.
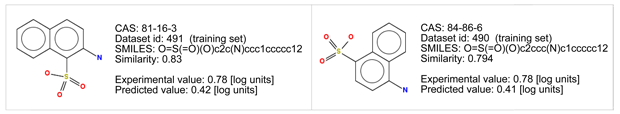
- The aromatic nucleus in the similar compounds has two fused rings, which increases logP and BCF. Thus, the target compound should have a lower BCF value. This is the case.
- Figure 1 shows that the target chemical is within the typical relationship between logP and BCF.
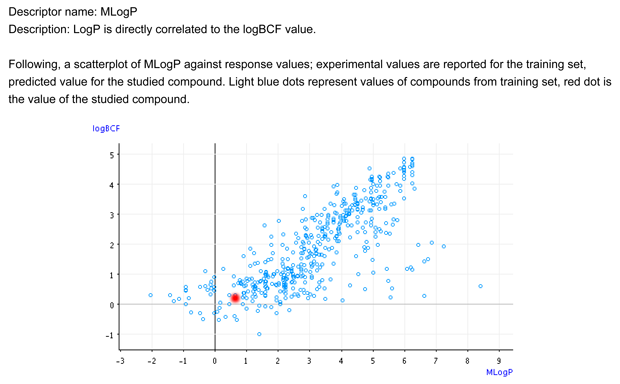
- Figure 2 shows that the BCF and logP values of the target compound are close and lower than the values of the similar compounds.
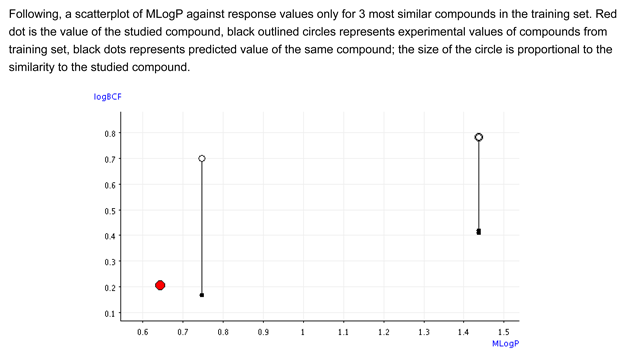
The predicted value is not inside the range defined by the logP and BCF values of the similar compounds, which represents the ideal situation. However, the values of the similar compounds can be used as safety margin, because the similar compounds are very probably with higher BCF values, because they have two aromatic rings. Thus, the evaluation is still acceptable
- Figure 2 shows that the predictive model has the tendency to underestimate the BCF value. It is likely that the real BCF value is higher than the predicited one, but in any case it should be lower than 0.78, which is the value for the analogs with the naphthalenic structure.
Not Reliable Evaluation
The VEGA evaluation of the applicability domain is not good, and a grey or a pale green button appears in the summary evaluation
This means that VEGA found one or more critical behavior.
VEGA is set to be quite conservative, in the Examples 1 and 2 the user should analyze independently the results and reach a conclusion that the prediction is anyhow reliable, on the basis of the information given by the tools for read across.
On the other hand, in the Examples 3 and 4, even analyzing independently the read across results a large uncertainty remains, and it is not possible to get a reliable conclusion. The user should use other models, such as T.E.S.T. or EPISuite, and compare the results obtained with these other models. If there is a consensus, the result could be used. If there is no consensus, at least one model is not reliable.
Examples
NAME
2,3,7,8-Tetrachlorodibenzo-p-dioxin
CAS
1746-01-6
SMILES
O1c3cc(c(cc3(Oc2cc(c(cc12)Cl)Cl))Cl)Cl
PREDICTED VALUE
3.02
Model assessment
 Unable to provide a prediction due to the presence of one or more fragments related to model outliers. The following relevant fragments have been found: O linked to aromatic and 3 Br/Cl linked to aromatic (SO 05).
Unable to provide a prediction due to the presence of one or more fragments related to model outliers. The following relevant fragments have been found: O linked to aromatic and 3 Br/Cl linked to aromatic (SO 05).
Let’s consider the read across evaluation:
- There are chemicals with very good similarity. Look at the similarity values > 0.95, and at the chemical structures.
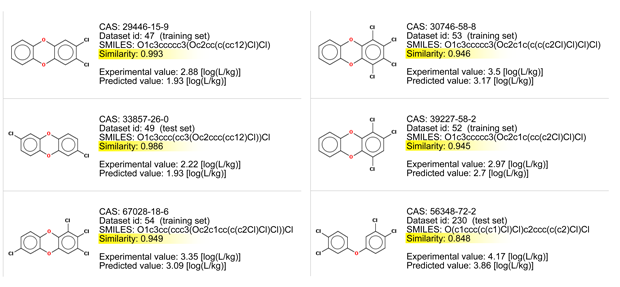
- All these chemicals differ only for the number or the position of the chlorine atoms.
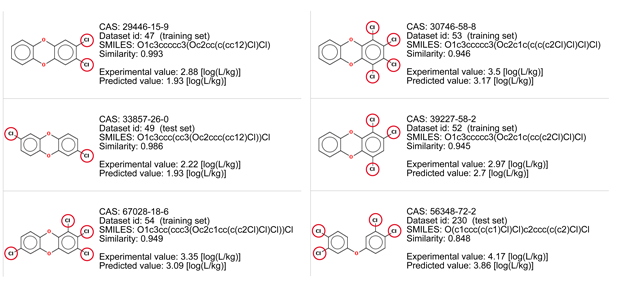 BCF values increase with the addiction of the chlorine atoms. The last compound is slightly different for the lack of an oxygen atom, and for this reason it should be more lipophilic and with a higher BCF value
BCF values increase with the addiction of the chlorine atoms. The last compound is slightly different for the lack of an oxygen atom, and for this reason it should be more lipophilic and with a higher BCF value
- Figure 1 shows that the target chemical is within the typical relationship between logP and BCF.
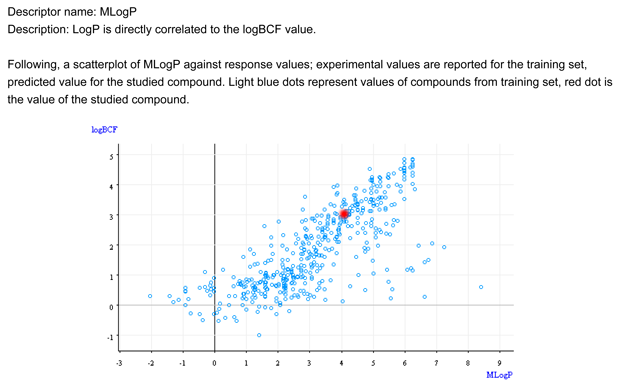
- Figure 2 shows that the BCF and logP values of the target compound are close to the values of the similar compounds. A trend appears.
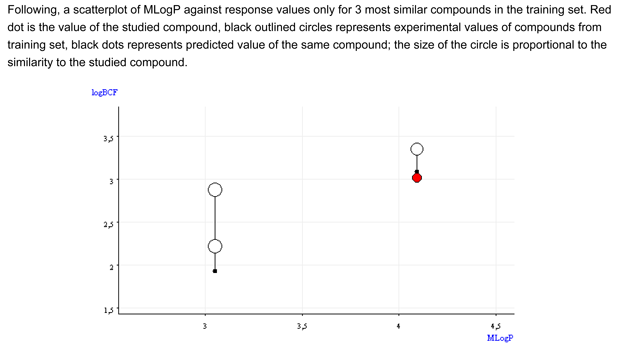
- The QSAR model underestimates BCF, of about 0.3-0.9 log units.
Thus, an increase of 0.4/0.5 should be considered to be safe. Remember that the typical uncertainty for experimental BCF values is about 0.4-0.5.
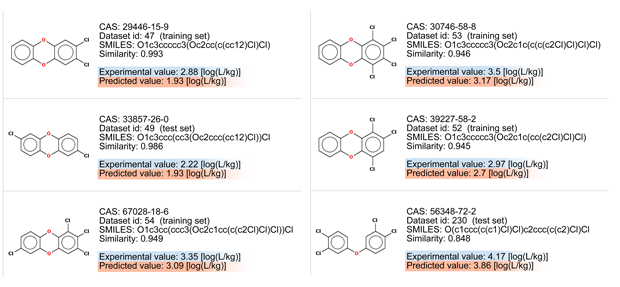
- The similar compounds with higher BCF values have four chlorine atoms.
Thus, the BCF value of the target compound is expected to be close to the BCF values of these two compounds (only one of them represented in Figure 2)
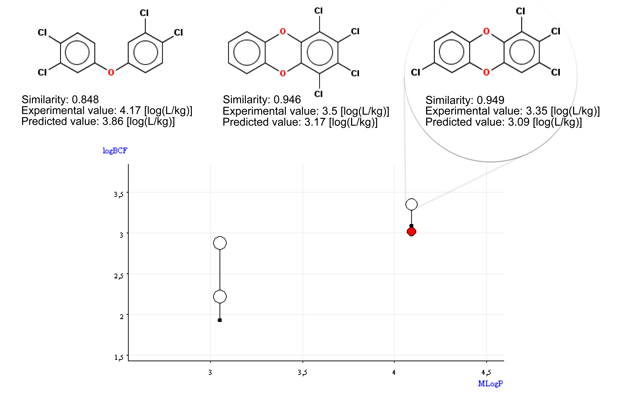
Conclusion: The compound is out of the applicability domain only for the presence of O linked to aromatic and 3 Br/Cl linked to aromatic, which is associated to higher uncertainty of the predictions; nevertheless, the read across assessment supports with a reasonable documentation that the QSAR prediction is realistic, even though probably slightly underestimated, considering the results on the other tetracholodibenzo-p-dioxins.
NAME
4-Aminophenol
CAS
123-30-8
SMILES
Oc1ccc(N)cc1
PREDICTED VALUE
0.47
Model assessment
 Model assessment: Prediction is logBCF = 0.47, but compound could be OUT of the applicability domain for the following reasons:
Model assessment: Prediction is logBCF = 0.47, but compound could be OUT of the applicability domain for the following reasons:
- accuracy of prediction for similar molecules found in the training set is not adequate;
- similar molecules found in the training set have experimental values that slightly disagree with the target compound predicted value;
- the maximum error in prediction of similar molecules found in the training set has a high value, considering the experimental variability.
The following relevant fragments have been found: OH group (PG06); NH2 group (PG 07).
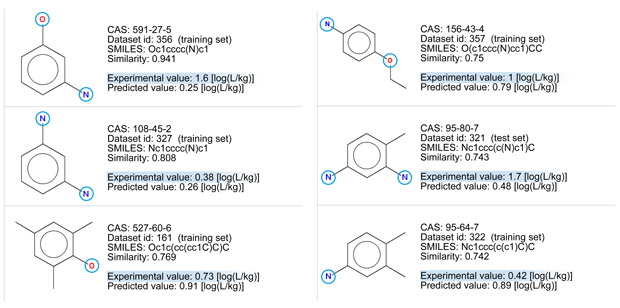
Let’s consider the read across evaluation:
- There are some chemicals with good similarity. The first compound differ only for the position of substituents. Look at the similarity values ≥ 0.75, and at the chemical structures.
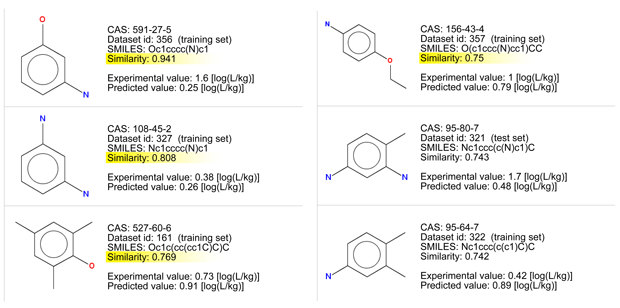
- All the chemicals have a low BCF value due to the presence of polar groups.
- Figure 1 shows that the target chemical is within the typical relationship between logP and BCF.
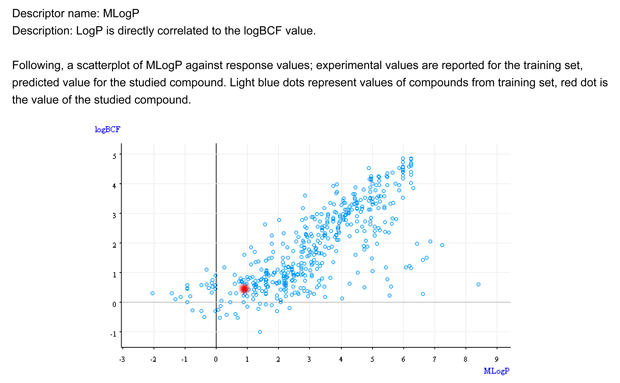
- Figure 2 shows that the BCF and logP values of the target compound are close the values of the similar compounds.
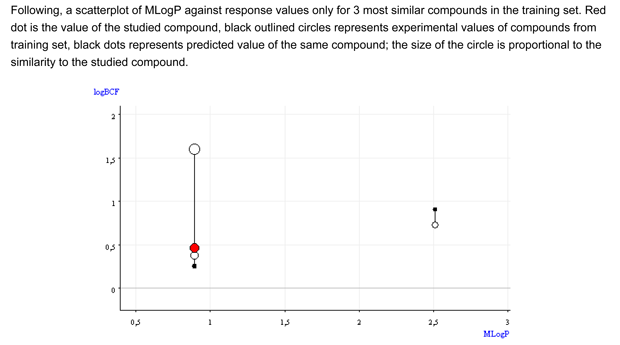
Conclusion: The low applicability domain index value is associated to the high uncertainty of the predictions; nevertheless, the read across assessment supports with a reasonable documentation that the target compound is nB. The read across value is higher than the predicted one, and close to 1.6, the value of the analog with substituents in meta. The compound number 4 in the "Applicability domain: Similar compounds, with predicted and experimental values" section, which is identical to the target compound but with an ethoxy group instead than the hydroxy group, has a BCF value of 1. This analog should have a BCF value higher than that of the target compound. Thus, the chemical is very probably non bioaccumulative, with a BCF value of 1.6, or lower.
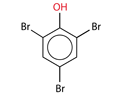
NAME
2,4,6-Tribromophenol
CAS
118-79-6
SMILES
Oc1c(cc(cc1Br)Br)Br
PREDICTED VALUE
0.81
Model assessment
Model assessment: Model assessment: Unable to provide a prediction due to the presence of one or more fragments related to model outliers.
The following relevant fragments have been found: O linked to aromatic and 3 Br/Cl linked to aromatic (SO 05); OH group (PG 06).
Let’s consider the read across evaluation:
- The chemicals are very similar; however, there are several chlorinated compounds with the same groups, and only one compound with a bromine atom.
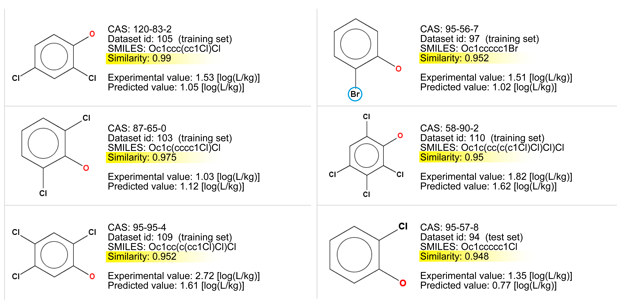
- Among similar compounds it seems that the BCF value increases with the addiction of chlorine atoms, even if the fifth compound, with four chlorine atoms, has a BCF value lower than the third one with three chlorine atoms. Similarly, the chemical with one chlorine atom (the sixth one) has a BCF value higher that that with two atoms (the second one).
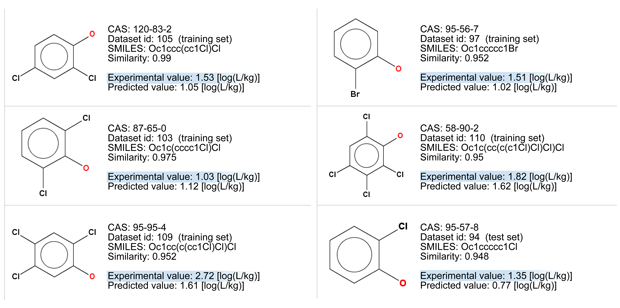
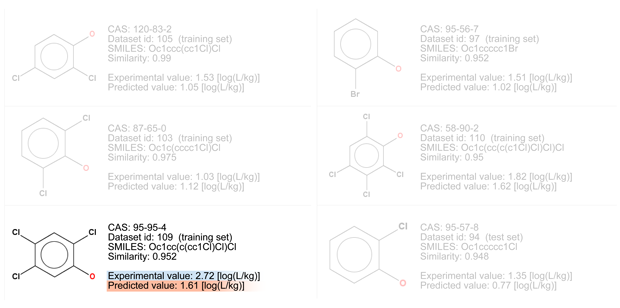
- Compounds with bromine atoms should be more lipophile than similar compounds with chlorine atoms, so it’s expected that BCF value of the target compound is higher than the predicted one, also considering the experimental value of the analog with three chlorine atoms (similar number 3: BCF = 2.72).
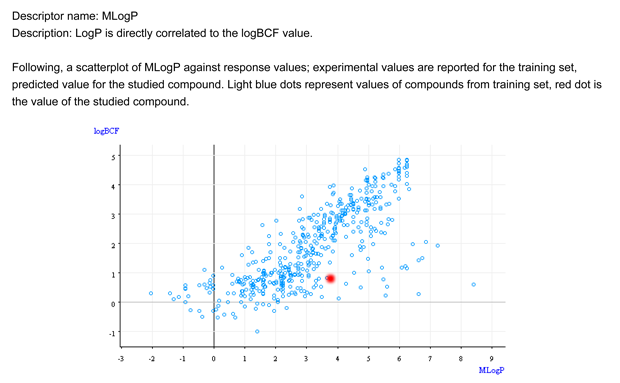
- Figure 1 shows that the chemical is not within the general relationship logP/BCF. The target compound is in the outliers area.
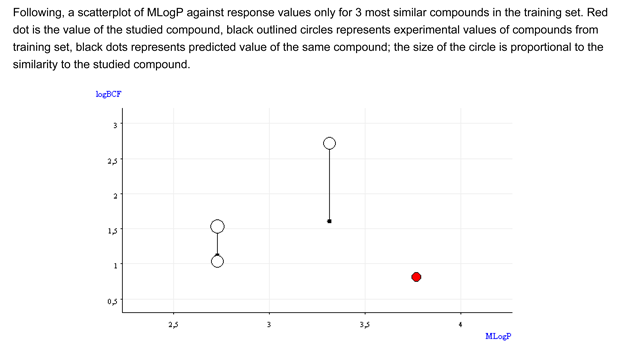
- Figure 2 shows that the BCF and logP values of the target compound are outside the range of the similar compounds. The predicted BCF value for the target compound does not follows the trend of the similar compounds.
Conclusion: The QSAR result for the target compound does not seems reliable. On the basis of the analysis of the similar compounds, the target chemical seems to be nB, but a large uncertainty remains, and it is possible that the target compound is B.
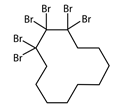
NAME
Hexabromocyclododecane
CAS
25637-99-4
SMILES
C1CCCCC(C(C(CCCC1)(Br)Br)(Br)Br)(Br)Br
PREDICTED VALUE
1.57
Model assessment
Model assessment: Unable to provide a prediction, compound is OUT of the applicability domain for the following reasons:
- accuracy of prediction for similar molecules found in the training set is not adequate;
- similar molecules found in the training set have experimental values that slightly disagree with the target compound predicted value;
- the maximum error in prediction of similar molecules found in the training set has a high value, considering the experimental variability;
- a prominent number of atom centered fragments of the compound have not been found in the compounds of the training set or are rare fragments.
Let’s consider the read across evaluation:
- The measured similarity is good, but the large ring of the target compound is quite peculiar and not present in the similar, simpler compounds.
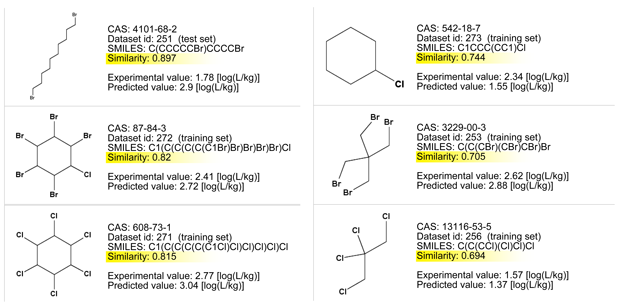
- Compounds with less bromine or carbon atoms have experimental values higher than the predicted value of the target compound.
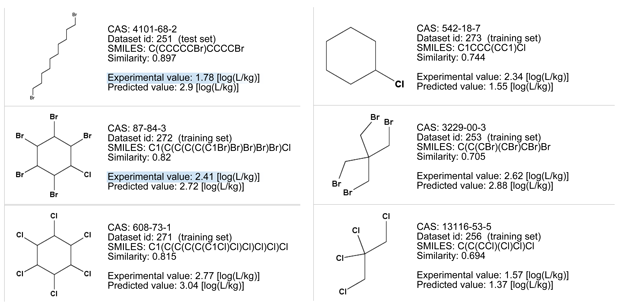
- All the similar chemicals are non-Bioaccumulative (nB) for the PBT assessment since their experimental BCF is lower than 3.3).
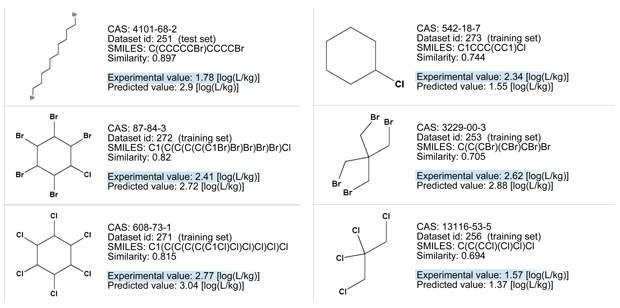
- Figure 1 shows that the chemical is not within the general relationship logP/BCF. It is an outlier.
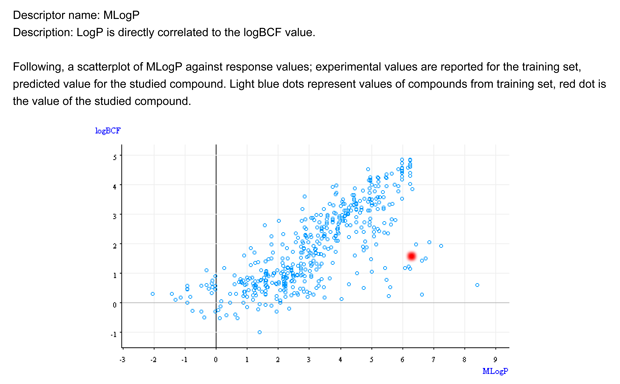
- Figure 2 shows that the BCF and logP values of the target compound are outside the range of the similar compounds.
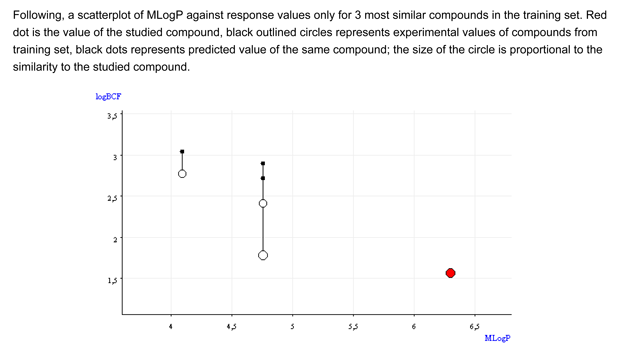
Conclusion: The QSAR result for the target compound does not seems reliable. On the basis of the analysis of the similar compounds, it is difficult to assign a BCF value.